 Auteurs: Massimo Nespolo et Benoît Guillot
Auteurs: Massimo Nespolo et Benoît Guillot
Université de Lorraine, CNRS, CRM2, Nancy
Téléchargement du logiciel CHARDI2015
Introduction et principe du calcul.
Les structures non-moléculaires peuvent normalement être décrites comme construites sur des polyèdres de coordination qui partagent des sommets, d’arêtes ou (plus rarement) des faces. Ces polyèdres sont typiquement centrés par des atomes électropositifs et présentent des atomes électronégatifs à leurs sommets, mais des exemples de structures où ces rôles sont inversés sont aussi connus. Il en résulte un réseau de liaisons chimiques qui peut être idéalement décrit comme un graphe dans lequel les nœuds représentent les positions atomiques et les arêtes des liaisons. Malgré sa simplicité, cette approche de type Madelung (basé sur des charges ponctuelles) peut fournir des informations structurelles significatives sur la base de la géométrie des polyèdres de coordination. Cette analyse est réalisée à travers la méthode de distribution des charges (CHARDI).
L’origine de cette approche peut être retracée à l’idée de force de liaison de Pauling, généralisée par la théorie de la valence de liaison (Bond Valence) afin de prendre en compte l’effet des différentes longueurs de liaison. La méthode de la valence de liaison utilise des paramètres empiriques dont les valeurs sont loin de faire l’unanimité, comme le montre le fait que des nombreuses avec des nouvelles valeurs continuent de sortir même après plus d’un demi-siècle de la présentation de la méthode.
Au contraire, CHARDI utilise simplement les distances de liaison expérimentales d’une manière auto-cohérente pour assigner à chaque liaison une « force » géométriquement définie (le poids de liaison). La somme des poids de liaison définit le numéro de coordination efficace (ECoN), un paramètre qui tient compte à la fois du nombre d’atomes coordonnés et de l’importance relative de chacun d’eux dans la coordination.
Les « charges » (nombres d’oxydation formels) sont ensuite distribués entre les atomes liés en fonction de ECoN et les valeurs résultantes sont sommées autour de chaque atome. Ceci conduit à des « charges » calculées qui donnent une estimation de la qualité du modèle de structure (pour les atomes au centre des polyèdres) et des sur-liaisons ou sous-liaisons, qui témoignant de l’existence de déformations structurelles (pour les atomes aux sommets des polyèdres).
Configuration requise et installation
CHARDI2015 fonctionne en environnement Windows et nécessite Redistribuable Visual C++ pour Visual Studio 2015, disponible gratuitement sur le site du Microsoft. Le logiciel (32 bits) est fourni avec un installeur qui contient toutes les librairies. L’espace disque occupé est d’une vingtaine de Mo environ.
 Lecture des données cristallographiques
Lecture des données cristallographiques
CHARDI2015 lit les fichiers CIF mais nécessite la présence des instructions
loop_
_atom_type_symbol
_atom_type_oxidation_number
suivies du symbole de l’atome et de son nombre d’oxydation. Ces données ne sont pas forcément présentes dans les fichiers CIF obtenus par des bases des données (ICSD, OCD etc.) : elles doivent alors soit être ajoutées à la main, soit à travers un fichier texte additionnel, comme expliqué dans le manuel de l’utilisateur.
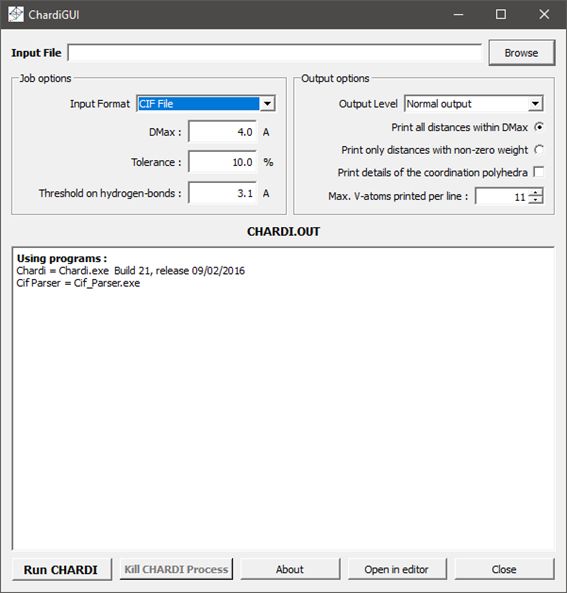
La lecture du fichier CIF est réalisée à travers une interface graphique montrée en Figure 1.
Le fichier CIF lu à l’aide du bouton « Browse » va être analysé et les informations nécessaires au calcul extraites dans un fichier intermédiaire qui sera ensuite utilisé pour le calcul lui-même. En cas de problème dans la conversion du fichier CIF une alerte apparaît dans la fenêtre du logiciel, avec renvoi vers un fichier « log » qui détaille le processus de conversion et les erreurs trouvées. Si non, les résultats du calcul apparaissent dans la même fenêtre et peuvent aussi être visualisés avec un éditeur de texte en cliquant sur le bouton « Open in editor ».
Exemple
Charge Distribution (CHARDI) et Bond Valence Sum (BVS) pour la structure de KNaTiO3 (Werthmann and Hoppe, 1985). CHARDI calculé avec CHARDI2015 (Nespolo and Guillot, 2016); BVS computed with Kdist (Knizek, 2017).
|
|
CHARDI
|
BVS
|
|
Ti
|
4.01
|
3.92
|
|
K
|
0.99
|
0.83
|
|
Na
|
1.01
|
1.09
|
|
O1
|
-2.06
|
-1.95
|
|
O2
|
-1.97
|
-1.94
|
Références
Knizek, K. (2017). KDist, version 4.31. part of the Kalvados software suite (http://www.fzu.cz/~knizek/kalvados). Institute of Physics, ASCR, Praha, Czech Republic.
Nespolo, M., (2016). Charge Distribution as a tool to investigate structural details. IV. A new route to heteroligand polyhedra. Acta Crystallographica B72, 51-66.
Nespolo, M., Guillot, B. CHARDI2015: Charge Distribution analysis of non-molecular structures. J. Appl. Crystallogr., 49, 317-321.
Werthmann, R. and Hoppe, R. (1985). Ein neues einfaches Titanat: KNaTiO3. Z. Anorg. Allg. Chem., 523 54-62.