 |
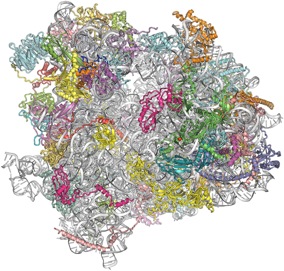
Marat Yusupov et Adam Ben Shem (IGBMC, Strasbourg) commentent la résolution de la structure du ribosome 80S eucaryote, qu’ils ont publiée dans Science en Décembre 2011 |
Ribosomes translate the information encoded in messenger RNA and synthesize proteins in all living organisms. Marat Yusupov and his team at the Institut de Génétique et de Biologie Moléculaire (Strasbourg) solved the first X-ray crystallography structure of an eukaryotic ribosome (yeast 80S ribosome) at medium resolution in 2010, and have now pushed it to 3 Å resolution (Science, 2011). This is the first detailed description of this huge cellular machine, comprised of 79 proteins and about 5600 nucleotides. It uncovers for the first time the eukaryotic-specific elements (representing 1,3 MDa) and how they interact with the universally conserved ribosomal core (2MDa), opening future avenues for therapeutic strategies. This is also the largest asymmetric content ever solved in biology, which raised many X-ray crystallography challenges. Marat Yusupov and Adam Ben Shem comment on how they cracked this seminal structure.
[AFC] Your previous 80S ribosome structure was at solved at 4.15Å resolution, which did not allow to see protein details. How did you extend the resolution to 3Å?
[Adam Ben Shem and Marat Yusupov] First, instead of collecting data on a single crystal, we used a large number of crystals. We also refined the dehydration procedure, notably making the stepwise increase in the concentration of PEG 6000 in the mother liquor more robust and reproducible. Improvements in the data collection strategy were also crucial. Previously we used the beam at maximal flux with exposure times of a few seconds per frame which resulted in very fast deterioration of the crystals due to radiation damage.We now follow a new strategy, recently developed in the SLS synchrotron, which takes full advantage of the unique features of the single photon counting pixel detector PILATUS 6M. The beam was attenuated to less than 10% of the maximal flux, exposure time was reduced to one second and oscillation step was 0.1 degree . As a result each exposure spot in each crystal "survived" on the beam for several hundred frames. We thus obtained tens of thousands of frames (13 crystals, multiple spots per crystal) that gave a complete data-set with very high redundancy. Although outer shell diffraction spots in individual frames were very weak/noisy, averaging over the highly redundant data-set yielded an improvement of roughly 0.5-0.6A in resolution over the conventional data-collection method.
[AFC] The final structure comprises two full ribosomes, that is over 400000 atoms to build and refine. This must have been a tremendous effort to get it done.
[ABS and MY] Building and refining the atomic model has kept three of us (Adam Ben Shem, Nicolas Garreau de Loubresse and Sergey Melnikov) in the darkness of the graphics room for a year. The structure was solved by combining our previous 4.15Å model with SAD phases derived from over 1300 osmium hexamide sites. Building posed a challenge as the location of many of the eukaryotic ribosomal proteins was unknown. Proteins with no homologues in prokaryotic ribosomal structures were first identified according to their size, fold, unique features such as a zinc finger and finally by the fit between their sequence and the electron density. 79 proteins were eventually identified, including an unexpected extra non-ribosomal protein. We discovered a striking abundance of eukaryotic elements on the surface of the particle, while universal elements are mostly in the inside of the particle.
The structure of the eukaryotic ribosome at 3.0 Å resolution. Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. Science. 2011 (334)1524 [Pubmed]
Rédaction: Jacqueline Cherfils