Auteurs: Théo Leduc, Vedran Vucovic, Eva Mocchetti, Christian Jelsch &
![]() Téléchargement du logiciel MoProViewer (avec MoProSuite)
Téléchargement du logiciel MoProViewer (avec MoProSuite)
Plateformes: Windows 10, bientôt Linux
Navigation: Introduction/Visualisation/Suivi affinement/Propriétes/Références
1. Introduction
MoProViewer [1] est le logiciel de visualisation moléculaire associé au programme MoProSuite [2], spécialisé dans l’affinement de structures cristallines ou de modèles de densité électronique à partir de données de diffraction des rayons X à haute résolution. La MoProSuite est constituée de deux modules principaux : MoPro, pour l’affinement cristallographique et VMoPro, destiné au calcul de propriétés dérivées de la distribution de charge. Au départ, MoProViewer a été conçu au laboratoire CRM2 pour permettre à un utilisateur de MoProSuite de configurer graphiquement un calcul par le module VMoPro. Depuis, de nombreuses fonctionnalités ont été implémentées et continuent de l’être à un rythme régulier, permettant d’utiliser MoProViewer indépendamment de MoProSuite. MoProViewer est développé dans l’objectif de permettre l’exploration de propriétés liées à la distribution de charge moléculaire en proposant une expérience utilisateur agréable et intuitive.
Navigation: Introduction/Visualisation/Suivi affinement/Propriétes/Références
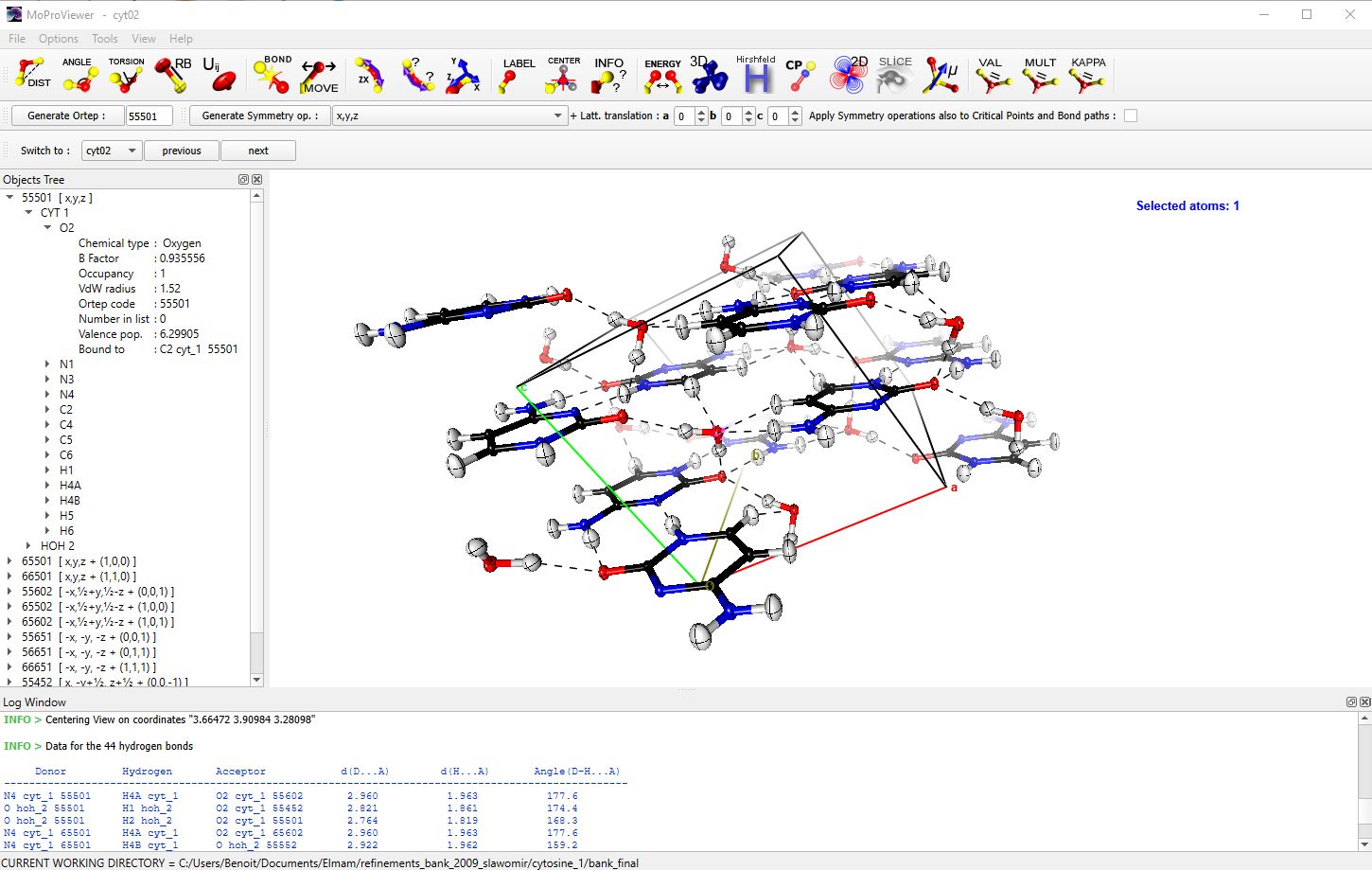
2. Visualisation de molécules et de structures cristallines
MoProViewer dispose de toutes les fonctionnalités attendues pour la représentation et l’étude de structures cristallines :
- Gestion des opérations de symétrie.
- Mesures de stéréochimie.
- Représentation des ellipsoïdes d’agitation thermique.
- Détection et représentation des liaisons hydrogène.
- Affichage des labels des atomes
- Etc
Navigation: Introduction/Visualisation/Suivi affinement/Propriétes/Références
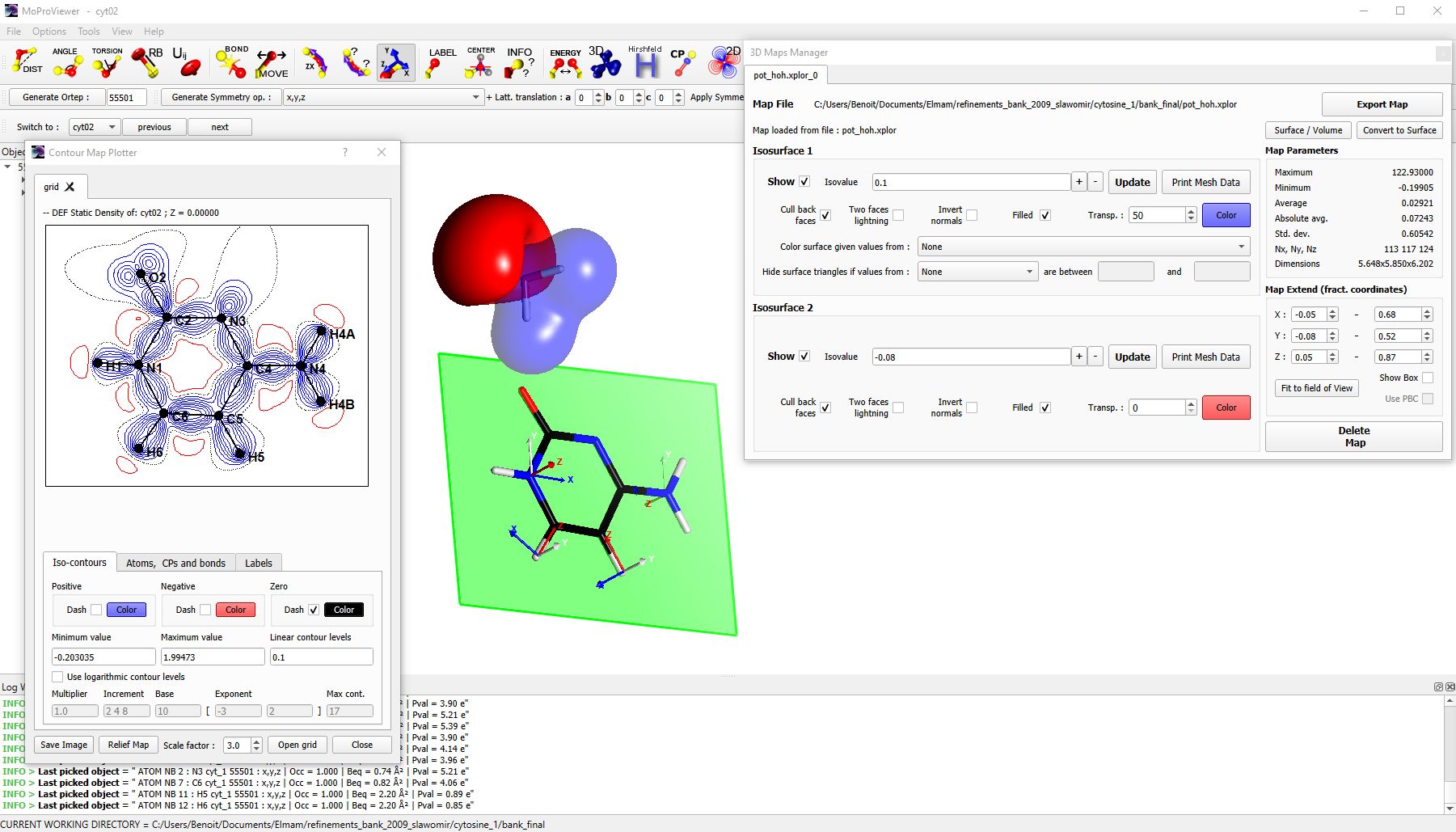
3. Suivi d’un affinement de densité électronique réalisé avec la MoProSuite.
MoProViewer peut être considéré comme une interface graphique pour VMoPro. En quelques clics de souris, sur la base de la représentation de la molécule en cours d’affinement, l’utilisateur peut demander dans MoProViewer le calcul par VMoPro de n’importe quelle propriété qui lui est accessible. MoProViewer ensuite affiche le résultat du calcul. Les propriétés disponibles via VMoPro sont très nombreuses, allant de diverses densités électroniques (totale, de déformation, de valence etc...) aux propriétés dérivées telles que le potentiel électrostatique, le Laplacien de la densité électronique, l’énergie d’interaction électrostatique etc... Les modes de représentation dans MoProViewer sont soit en 2D (cartes en niveau de contour) ou en 3D (iso-surfaces de propriétés). MoProViewer dispose d’un grand nombre de fonctionnalités en termes de configuration de ces représentations (figures 2, 3, 4).
Navigation: Introduction/Visualisation/Suivi affinement/Propriétes/Références
4. Propriétés dérivées de la distribution de charge
Outre son rôle d’interface pour le programme VMoPro, MoProViewer dispose d’un grand nombre de fonctionnalités qui lui permettent d’être utilisé de façon indépendante de la MoProSuite.
- Calcul et exploration de champs scalaires.
 |
 |
|
(a) |
(b) |
|
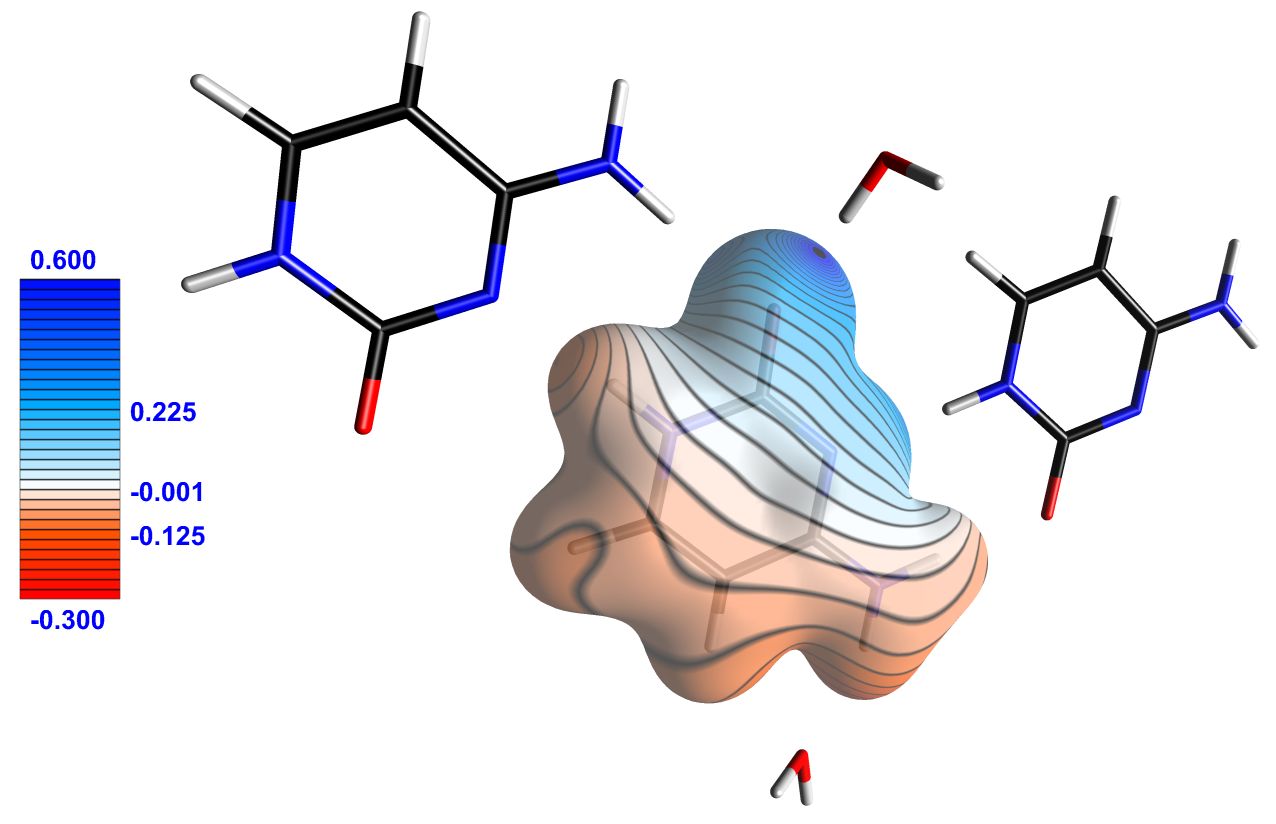
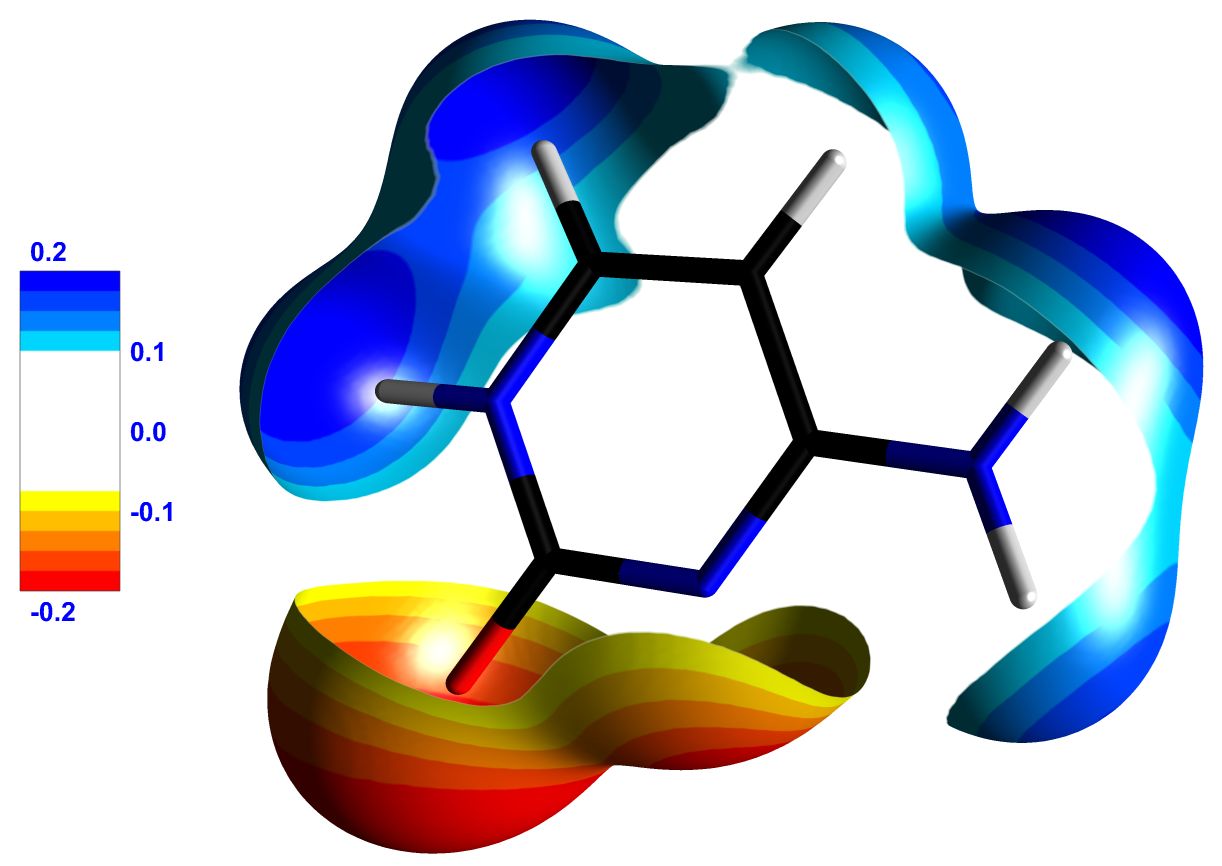
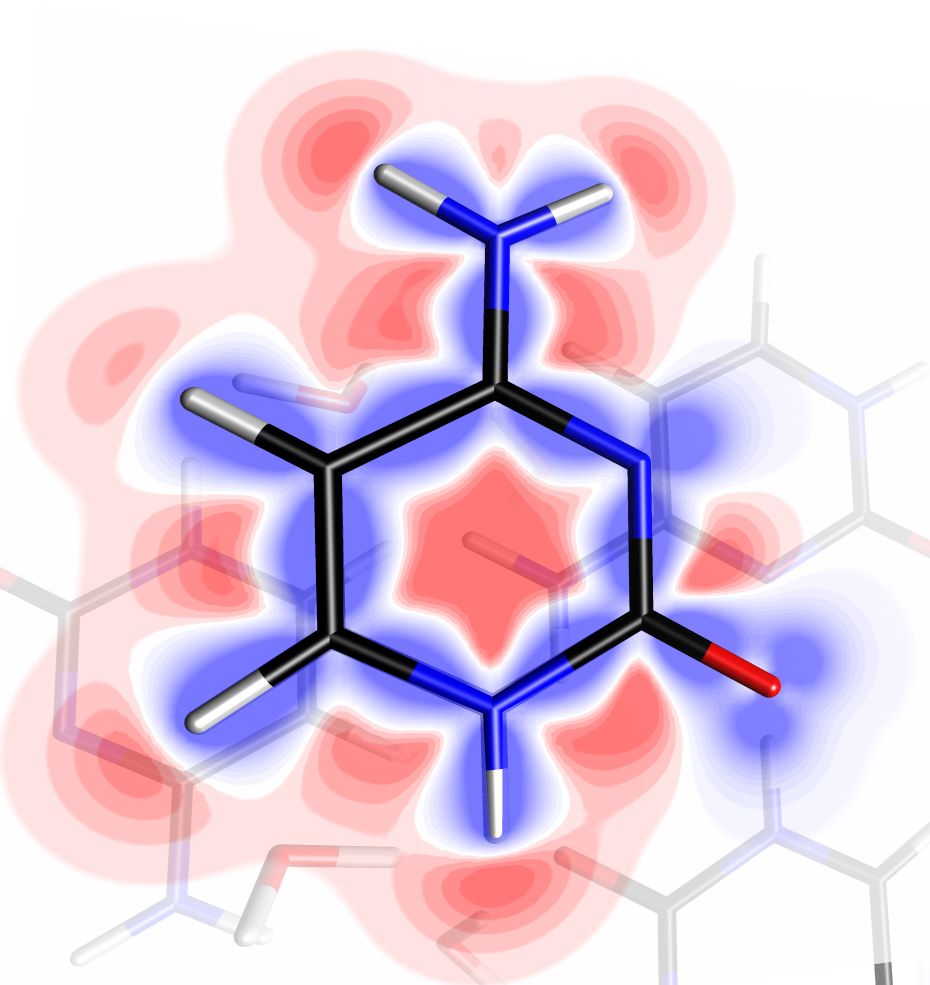
Figure 3 : (a) Molécule de cytosine représentée entourée d’une isosurface de 0.1 e/Å3 de densité électronique totale. La surface est colorée par le potentiel électrostatique généré par l’environnement cristallin. (b) Molécule de cytosine représentée entourée d’une isosurface de 0.05 e/Å3 de densité électronique totale, colorée par son propre potentiel électrostatique. Seules les régions de la surface où le potentiel est supérieur à 0.1 e/Å ou inférieur à -0.1 e/Å sont représentées, faisant apparaitre clairement ses sites électrophiles et nucléophiles. Les légendes portent sur les valeurs du potentiel électrostatique (en e/Å). Images exportées par MoProViewer. |
|
 |
 |
|
(a) |
(b) |
|
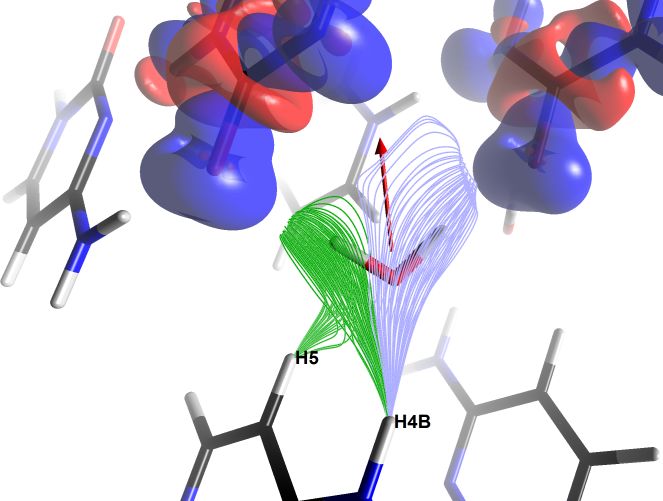
Figure 4 : (a) Densité électronique de déformation statique représentée pour une molécule de cytosine dans le cristal à l’aide de l’outil d’exploration de champs scalaires. La densité est projetée sur un plan de coupe translucide, négative en rouge et positive en bleu. Les contours visibles sont d’environ ±0.3 e/Å3. (b) Lignes de champ électrique auxquelles est soumise une molécule d’eau dans le cristal de cytosine monohydrate. Deux faisceaux de lignes sont représentés, créés à l’aide de deux instances de l’outil d’exploration de champ scalaire. Les lignes émanent des atomes H5 et H4B d’une molécule de cytosine, et convergent vers des paires d’électrons libres d’atomes d’oxygène des groupements carbonyle de deux autres molécules (dont les densités électroniques de déformation statiques sont représentées en isosurfaces translucides de valeurs ±0.05 e/Å3). On constate le relatif alignement des lignes de champ électrique avec le moment dipolaire de la molécule d’eau, représenté en rouge (échelle 3.3 Å/e̝Å, valeur 0.48 eÅ soit 2.3 D). Images exportées par MoProViewer. |
|
- Topologie de la densité électronique
 |
|
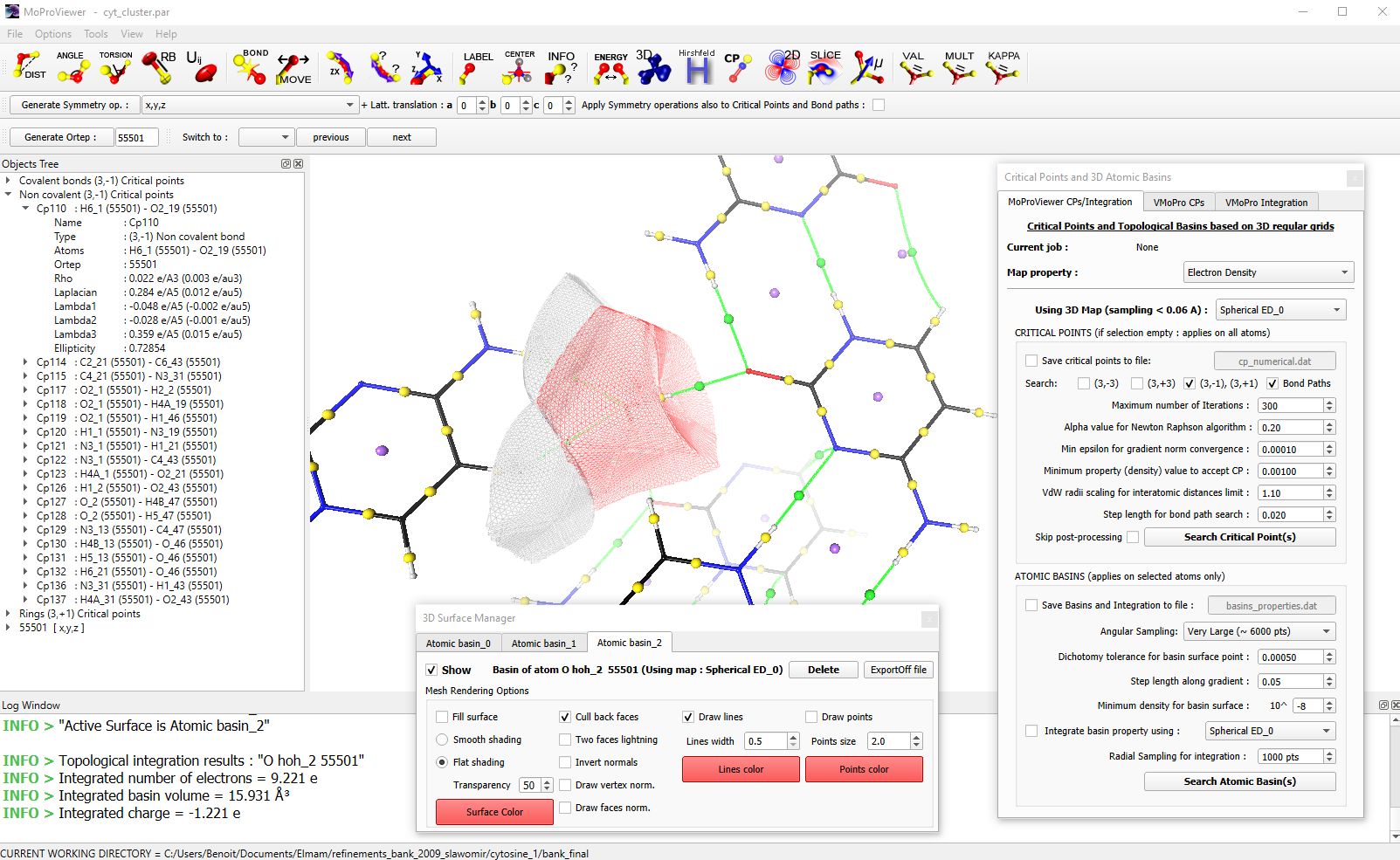
Figure 5 : Une portion du cristal de cytosine monohydrate et représentée. Les points critiques de liaisons covalentes sont représentés en jaune, ceux de liaisons hydrogène en verts, et les points critiques de cycle sont en bleu. Les points critiques apparaissent dans l’arborescence des objets visible à gauche. Trois bassins topologiques sont représentés en mode « wireframe » : ceux d’un atome d’oxygène d’une molécule d’eau (en rouge) et ceux de deux atomes d’hydrogène d’une molécule de cytosine voisine. Dans la fenêtre d’information (en bas) sont visibles les informations d’intégration de la densité électronique dans le bassin de l’atome d’oxygène. L’outil d’analyse topologique est visible à gauche, et l’outil de gestion des bassins atomiques est visible en bas. |
- Transfert et polarisation de la densité électronique
 |
|
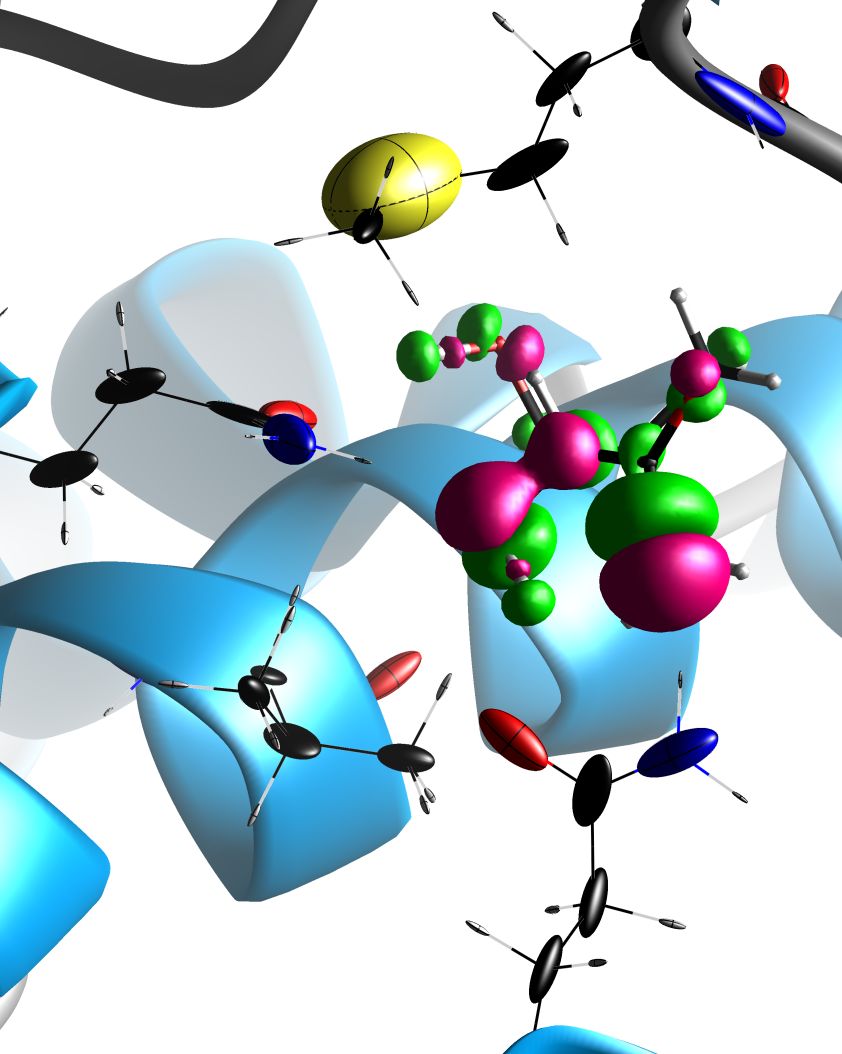
Figure 6 : un ligand de la protéine Panthotenate Synthetase (code pdb : 1N2J) est représenté dans son site de fixation, avec la densité électronique d’induction dipolaire montrée en isosurfaces de ±0.02 e/Å3. Cette densité électronique est représentée en rose là où le phénomène de polarisation a mené à une accumulation de densité électronique, et en vert dans le cas contraire. Les tenseurs de polarisabilité atomique des acides aminés à proximité du ligand sont représentés sous forme d’ellipsoïdes. |
- Calculs rapides d’énergies d’interaction électrostatique
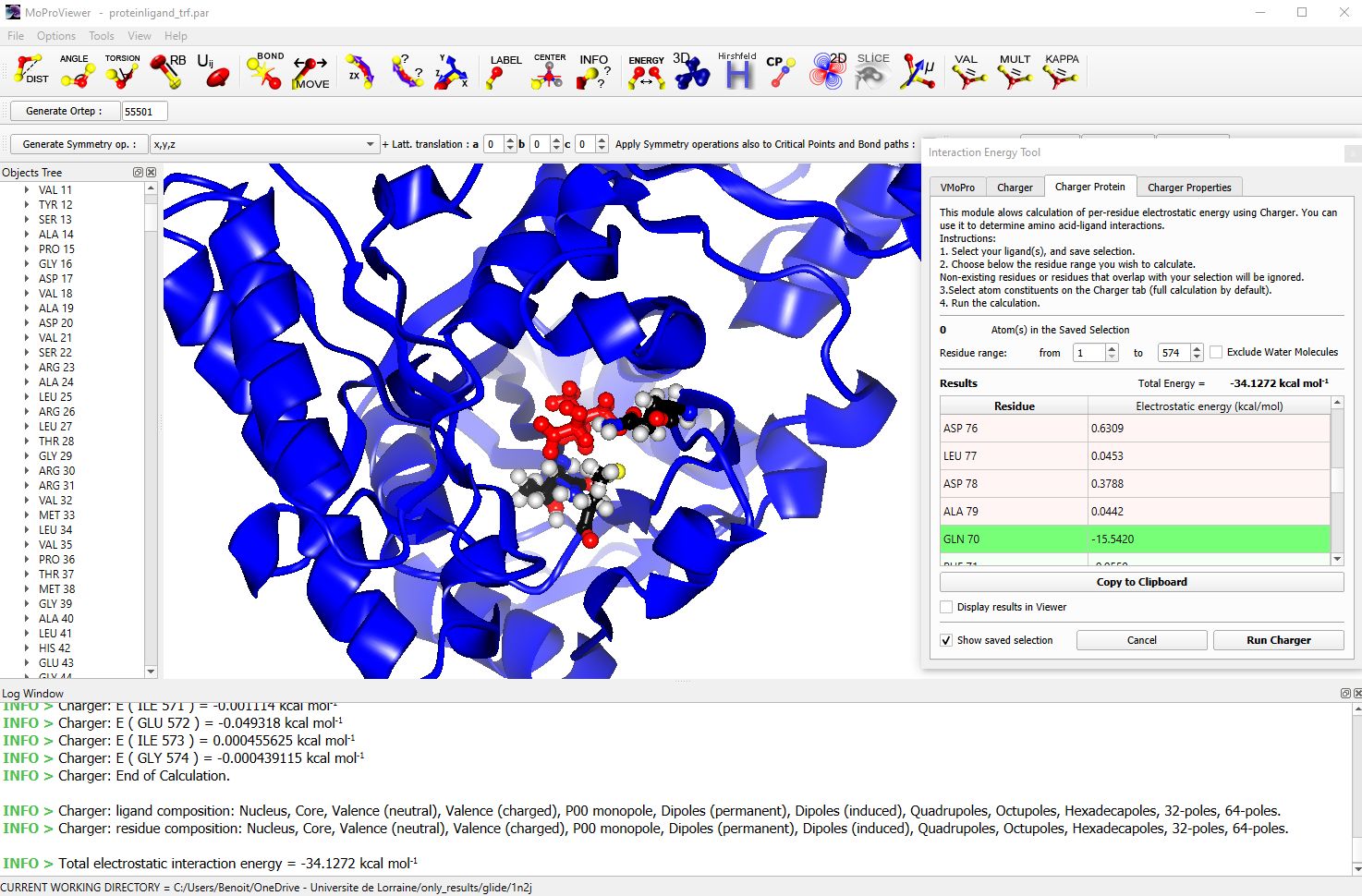 |
|
Figure 7 : La structure de la protéine Panthotenate Synthetase représentée en mode « cartoon ». Le ligand est en rouge et seuls les deux résidus qui contribuent le plus à la stabilisation électrostatique du ligand sont représentés. L’outil de calcul rapide des énergies d’interaction est visible à droite, avec en vert la contribution de GLN70. Le calcul des contributions des 574 acides aminés à l’énergie d’interaction électrostatique totale entre la protéine et le ligand prend une dizaine de secondes sur un Intel® Core i7 récent. |
Navigation: Introduction/Visualisation/Suivi affinement/Propriétes/Références
5. Références